About Angelman Syndrome
Genetics 201
For the basics of Angelman syndrome genetics, start here. If you've already read that page and want to dive deeper, you're in the right place.
Angelman syndrome (or AS) is caused by the lack of function of one specific gene, called UBE3A. To understand this, we first need to understand the basic structure and function of chromosomes, genes and proteins.
Generally, humans have 46 chromosomes, which come in 23 pairs. These chromosomes are inside most cells of their body, other than the egg or sperm, which only carries one copy, not a pair, and therefore they each have 23 chromosomes. We inherit one copy of each chromosome from the egg (maternal copy) and one copy from the sperm (paternal copy). Chromosomes are made up of a string of DNA, which are then categorized into separate groups, or genes, that each work independently to make proteins, which serves as the machinery which makes all of the cells of our body function properly. (See figure below)
The string of DNA within each gene creates an instruction manual that is made up of a long series of 4 letters, or basepairs, A (adenine), C (cytosine), G (guanine), and T (thymine), all in a different order based on the specific gene. This creates, or codes, for a story, which are the specific building blocks of proteins, called amino acids. This instruction manual generally must be read precisely, without any typographical errors, misspellings, letter deletions, or letter duplications for the end product, a protein, to be properly understood.
This is done through a few steps. One is that the DNA, which makes each gene, codes for a message that is more easily understood by the cell, called messenger RNA (mRNA). This message is then transported to protein factories which code for the protein that each cell needs to properly function. The gene impacted in AS, known as UBE3A, contains approximately 2700 basepairs of DNA, which codes for a protein that contains approximately 875 amino acids.
This is a karyotype, or map, of the human chromosomes.
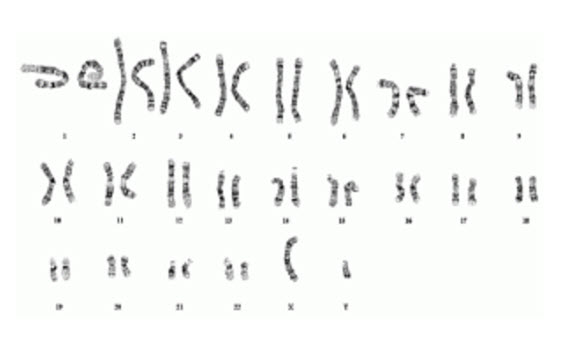
In this karyotype, you can see each set of chromosomes matched up from biggest (chromosome 1) to smallest (chromosome 22), and then the 2 sex chromosomes, in this case in a male (XY) or a female would be XX. Within a pair of chromosomes, you will have two copies of a gene that differ from each other in small ways because one came from the maternal side and one came from the paternal side, which are not identical. These differences are called gene variants or “alleles”, which we also refer to as “maternal allele” and “paternal allele.” Most of the time these differences don’t change function of the proteins. They just make us all unique individuals.
Each chromosome contains a different string of DNA, which is separated into different genes. There are over 20,000 genes in the genome, which means if we count maternal and paternal copies, there are over 40,000 genes—many of which are duplicated! We are going to focus on a single gene, which lives on chromosome 15, called the UBE3A gene, which is the causative gene in AS.
Angelman Syndrome was first described in 1965 by Dr. Harry Angelman based on the described common traits of individuals he was seeing in his clinical practice. It was not until 1987 that this condition was recognized to be due to a defect on chromosome 15. In 1997 the gene UBE3A was identified to be the causative gene leading to AS, making precision therapeutic development possible.
Generally, the UBE3A gene is present on each copy of chromosome 15 in the pair; the maternal allele and the paternal allele. The vast majority of cells in the body make UBE3A protein using the instructions from this gene, provided by both alleles. However, in neurons, this is different. It just so happens that in neurons, the cells of the brain and spinal cord, the instruction to read UBE3A is silenced on the paternal allele and is active on the maternal allele. This is a very unique phenomenon called “imprinting,” which is a form of gene regulation by the silencing of one parental copy. This is typically the case for all humans, where, in neurons, the maternal copy of UBE3A is expressed but the paternal copy of UBE3A is silenced, and therefore only the maternal copy is producing the needed UBE3A protein. All other cells of the body have both copies producing this protein.
This silencing mechanism occurs due to the presence of a strand of nucleic acids that is RNA, produced from the DNA, of the paternal chromosome. This strand is known as the “antisense transcript (ATS)” or UBE3A-ATS. (See figure below: Modified from N. Khatri et al. Front. Mol. Neurosci. 2019)). This is also sometimes referred to as a long non-coding strand of RNA (lnc-RNA). The term “antisense” is used because the instructions for the UBE3A-ATS are produced by DNA instructions that are read in the opposite direction as those that would produce UBE3A.
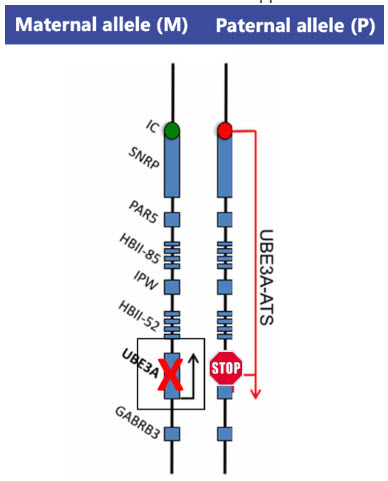
Notice the paternal copy of the gene is silenced from a STOP that is caused by the UBE3A-ATS, while at the same time, in AS, the maternal copy of the UBE3A gene is generally not functioning, or missing.
Production of the UBE3A-ATS prevents the reading of the paternal UBE3A gene on the paternal allele, leaving only the maternal copy active. Exactly how the production of the UBE3A-ATS blocks this reading of the paternal UBE3A is still being studied, but we know we can manipulate it.
Therefore, if there is a problem with the UBE3A gene on the maternal allele, then neurons do not have the ability to make the UBE3A protein from either copy, making neurons essentially deficient. This deficiency in maternal UBE3A expression results in the condition of AS.
There are 5 main ways in which the genetics can cause maternal UBE3A deficiency, which is shown here in this schematic.
This illustration depicts a schematic of the 15th chromosome and the different mechanisms, or genotypes, that can lead to AS. The “M” or “Maternal” allele represents the chromosome inherited from the egg while “P” or “Paternal” represents that inherited from the sperm. The orange line is the region that codes for the UBE3A gene. The first example shows a typical pair with the father’s UBE3A silenced, while the mother’s copy is active and UBE3A is expressed, making the genetic instructions in the neurons appropriate. The remaining examples illustrate the known mechanisms that cause AS by making the mother’s genetic information inaccessible to the cells in the brain. For example, in deletions, the UBE3A gene is completely missing, in UBE3A mutations the UBE3A gene is “misspelled” which makes this copy of UBE3A non-functional and in UPD and ICD there is double silencing of both alleles. Lastly, rarely, individuals have a mosaic genotype where some cells have maternal UBE3A expressed and others do not. In addition, there are some individuals have all or most of the symptoms of AS, but all genetic testing evaluated turned out normal. In many of these cases, it is likely that we just don’t yet have the technology and expertise to understand what type of AS they have as it is very rare, or the testing is dated and new testing with whole genome sequencing would give a more clear answer. This could help these families understand what is causing the symptoms in their loved one(s).
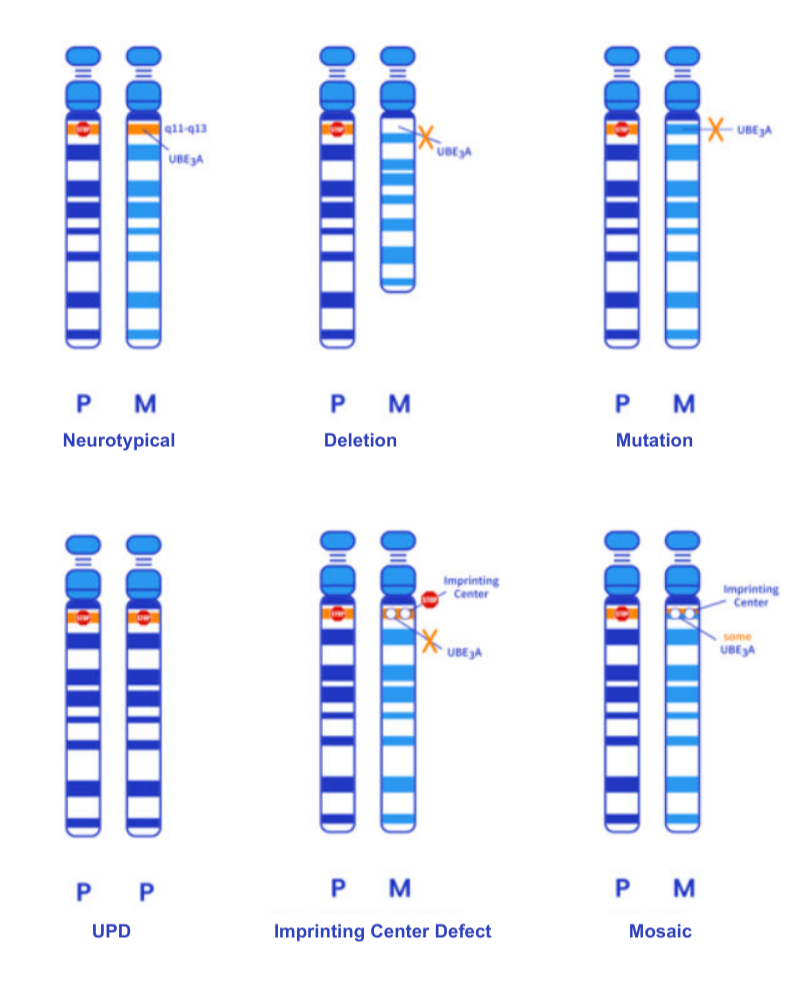
The different genetic “types” that cause AS are referred to as “genotypes” and we have 5 main genotypes.
Deletion
Mutation
Uniparental Disomy (UPD)
Imprinting Center Defect (ICD)
Mosaic
For more information on the different genotypes, see this page. For information on testing for them, go here.)
Most cases of AS are due to random events that occur during the development of the egg within the developing ovary, and are not the result of anything the mother did during the pregnancy of the child with AS. This event happened before getting pregnant, it acutally occurred before the mother was even born. . In the case of UPD, the fertilized egg may recognize that it only has one chromosome 15 and not a pair. This would be fatal to the embryo, so the egg copies the chromosome 15 that is present to make a pair and try and repair the defect. If the chromosome that is copied is the paternal chromosome, AS results because neither chromosome will transcribe UBE3A since both alleles are recognized as paternal. This again, does not happen because of anything the mother did or experienced during pregnancy, but in fact occurred at the time of fertilization at the single cell stage.
So, overall, regardless of the genetics behind AS, the commonality is that all individuals living with AS are missing the expression of UBE3A protein in neurons—commonly a random event. Regardless of the underlying cause, the loss of neuronal UBE3A expression results in a plethora of symptoms because the UBE3A protein is incredibly important in neurologic function. Despite the specific genotype, or type of AS, the symptoms are relatively consistent across them all.
The UBE3A protein has multiple functions, many of which are not yet fully understood. You can think of proteins as ‘machines’ that have a specific job to do.
The main job for the UBE3A protein is in a process called ubiquitination. UBE3A acts in a complex with other proteins to keep balance both inside and outside of the neuron. This is from its ability to “tag” other proteins in the cell to be either taken away for degradation and recycling or to be protected and kept around a bit longer. The tag itself is a small protein called ubiquitin (named so because of its abundance in cells). Since UBE3A adds, or ligates, ubiquitin to proteins, the UBE3A machine is referred to as an enzyme called ubiquitin ligase. Thus, in neurons lacking UBE3A, proteins can accumulate to high levels or become unstable to low levels, both of which have a negative effect on neuronal function and coordination resulting in neurons have excessive amount of excitation and limited amounts of inhibition, or calmness. This calmness we often refer to as “tonic inhibition,” and it’s something our brains need to coordinate movement, brain activity, sleep, and most other functions. This is impacted in AS.
UBE3A protein has other jobs we know of as well. Adding ubiquitin to a protein can also regulate how that protein works, instead of targeting it for recycling. So, UBE3A can tag other proteins and potentially make them more or less effective at their jobs depending on the needs of the cell. We also know UBE3A can work with other proteins to control the expression of other genes on other chromosomes, which may be independent of its work with ubiquitin. Ultimately, understanding the critical functions of UBE3A will help us understand why neurons function poorly in the absence of UBE3A, which is the ultimate cause of AS. Scientists and clinicians are working to correlate protein function to the symptoms of AS.
We all know that UBE3A is vital to how the brain develops and controls speech, movement and learning. The large impact UBE3A has in neurons as well as at the junction between neurons (synapse) explains why individuals living with AS have challenges coordinating their muscles for controlled verbal speech, coordinated gait, and prolonged focus. The neurons are overactive making it also challenging to sleep and making our loved ones predisposed to seizures.
By understanding the genetics and physiology of AS it is easier to understand how impactful the various approaches can be to developing therapeutics that have the potential to be profoundly impactful for those living with Angelman syndrome. To do that, we have broken this down into three main therapeutic pillars.
Pillar 1: Replace the missing maternal UBE3A gene or protein in, or around, neurons: this can be done through gene replacement therapy (e.g. AAV-GT, HSC-GT) or enzyme replacement therapy (ERT).
Pillar 2: Unsilence the paternal copy of UBE3A. This can be done via various different mechanisms, all aiming for the same result. (e.g. ASO, CRISPR, Artificial Transcription Factors, shRNA/miRNA).
Pillar 3: Improve the symptoms of AS by targeting the downstream impact of UBE3A loss like improving the coordination and communication between neurons, at the synapse.